Integration of immune-cell type deconvolution features and prior-knowledge networks of TFs-gene interactions to characterize potential cell states of the tumor microenvironment using bulk RNAseq data
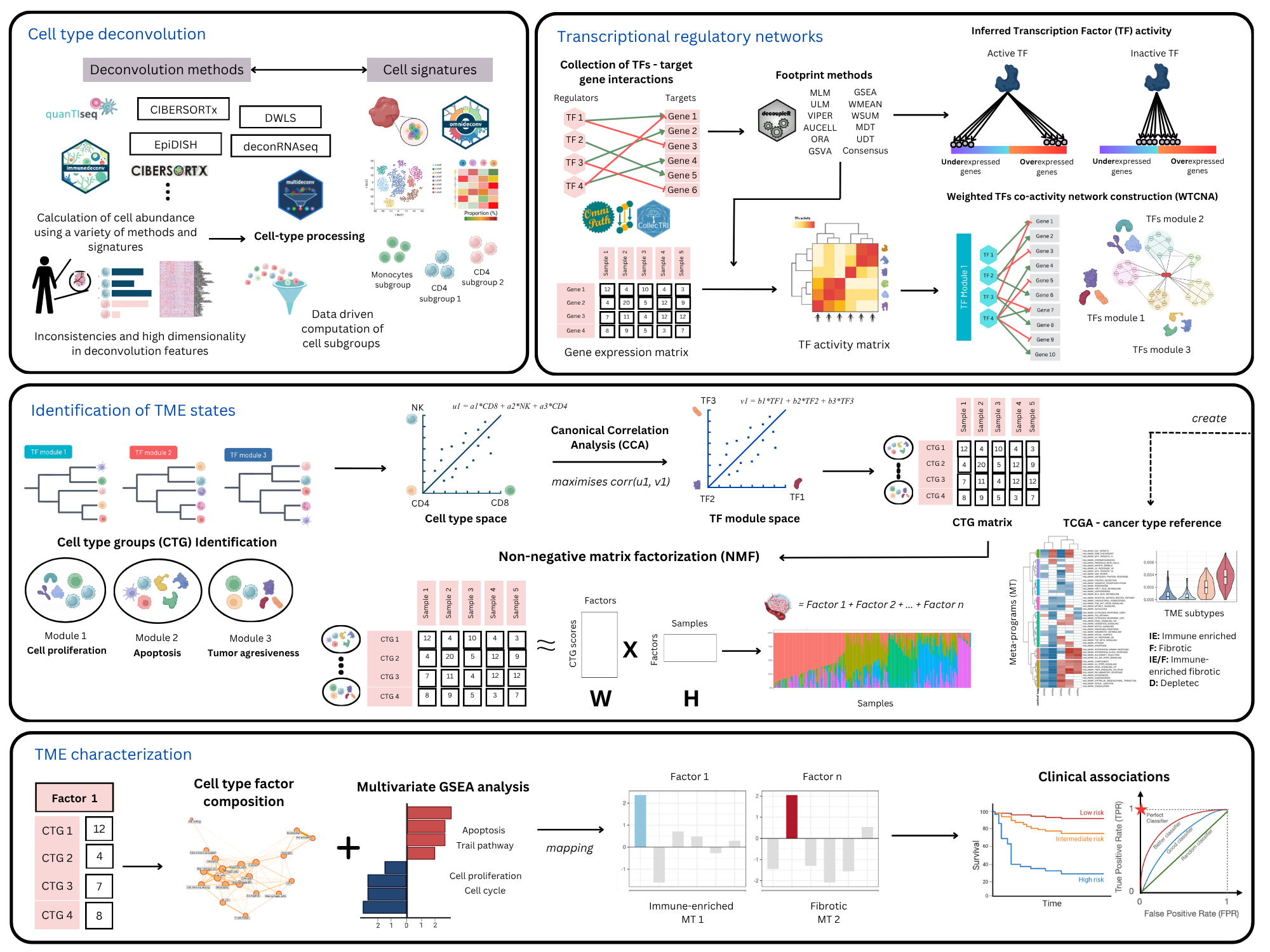
Figure 1. A schematic overview of the CellTFusion pipeline
Installation
To avoid GitHub API rate limit issues during installation, we recommend setting up GitHub authentication by creating and storing a Personal Access Token (PAT). You can do this with the following steps:
# install.packages(c("usethis", "gitcreds"))
usethis::create_github_token() #Create a Personal Access Token (if you don't have)
gitcreds::gitcreds_set() #Add the tokenYou can install the development version of CellTFusion from GitHub with:
# install.packages("pak")
pak::pkg_install("VeraPancaldiLab/CellTFusion")Launch the Shiny app
You can run the CellTFusion Shiny interface in two ways.
From an installed package:
library(CellTFusion)
shiny::runApp(system.file("shiny", package = "CellTFusion"))From this source repository:
shiny::runApp("inst/shiny")In the app, click Load tutorial data for a quick demo, then click Run CellTFusion. Results and downloadable files are written to your working directory.
General usage
These are basic examples which shows you how to use CellTFusion for different tasks. For a detailed tutorial, see Get started
Before running CellTFusion, make sure to set your working directory. The Results/ folder, where outputs will be saved, will be created in this directory.
setwd('~/path/to/directory')
library(CellTFusion)If you want to run the full pipeline in one step — including normalization, deconvolution, TF activity scoring, module construction, and pathway scoring — use the CellTFusion() wrapper function.
res <- CellTFusion(
raw.counts = raw.counts,
normalized = TRUE,
coldata = traitdata, # Optional metadata
deconv_methods = c("Quantiseq", "DeconRNASeq"), # Choose from Quantiseq, Epidish, DeconRNASeq, DWLS, CibersortX
cbsx.mail = "your_email", # Required if using CIBERSORTx
cbsx.token = "your_token", # Required if using CIBERSORTx
file_name = "TestRun",
min_targets_size = 15,
minMod = 20,
corr_mod = 0.25,
corr = 0.7,
pval = 0.05,
high_corr_groups = 0.85,
trait = "Best.Confirmed.Overall.Response", # Optional supervised analysis
trait.positive = "CR", # Define positive class for trait
return = TRUE
)NOTE: CIBERSORTx is included in the deconvolution methods, but it’s not an open-source program. To run it, please ask for a token in CIBERSORTx and once obtained, provided your username and password on the parameters credentials.mail and credentials.token.
Cell groups
You can use the construct_cell_groups() function to derive cell-type groupings based on transcription factor (TF) regulatory networks and deconvolution outputs. This supports both unsupervised and supervised analysis based on clinical traits.
# Run unsupervised cell group construction
cell_groups_unsupervised <- construct_cell_groups(
counts = counts_matrix, # gene expression (genes x samples)
tfs = tf_list, # list or matrix of transcription factors
deconv = deconv_matrix, # deconvolution results (samples x cell types)
network = tf_network_list, # TF networks for cell types
dt = deconv_subgroups, # deconvolution subgroup structures
clinical = clinical_data # clinical metadata
)
# Run supervised cell group construction based on a binary trait
cell_groups_supervised <- construct_cell_groups(
counts = counts_matrix,
tfs = tf_list,
deconv = deconv_matrix,
network = tf_network_list,
dt = deconv_subgroups,
clinical = clinical_data,
trait = "response", # binary trait column in clinical data
positive = "Responder" # class considered positive
)
# Output: both return a list with projected scores, composition, and loadings
# cell_groups$score: matrix of cell group scores (samples x groups)
# cell_groups$composition: cell types included in each group
# cell_groups$loadings: contribution of features per groupReplicate cell groups on an independent dataset
Use this function to apply the trained cell groups to an external test set and calculate group-specific composite scores.
test_scores <- compute.test.set(
deconv_res = deconv_res_test,
cell_groups = cell_groups,
features = selected_features,
deconvolution_test = deconv_matrix_test
)Issues
If you encounter any problems or have questions about the package, we encourage you to open an issue here. We’ll do our best to assist you!
Authors
CellTFusion was developed by Marcelo Hurtado in supervision of Vera Pancaldi and is part of the Pancaldi team. Currently, Marcelo is the primary maintainer of this package.